Het DNA bevat grote en kleine varianten, en deze worden niet allemaal even goed opgespoord. In deze introductie ga ik dieper in op de variatie in het DNA tussen twee personen, hoe we die kunnen opsporen en wat de gevolgen daarvan kunnen zijn.
De code van al wat leeft
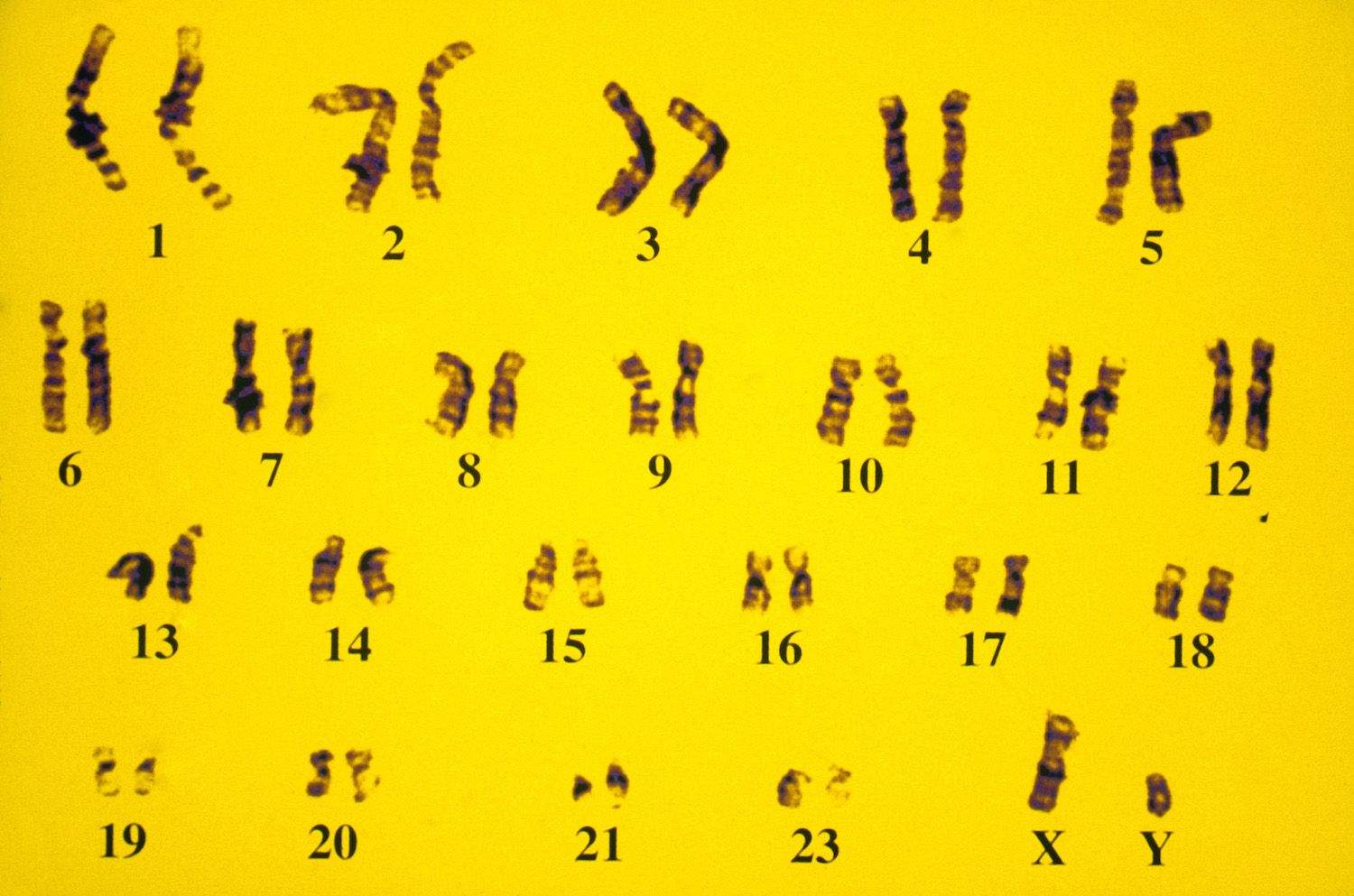
Ons erfelijk materiaal, het DNA, bestaat uit 47 stukjes: de chromosomen (Figuur 1). Die chromosomen bestaan uit zo'n 6.4 biljoen 'letters', de nucleotiden, letters die voorkomen in een A, T, G of C vorm. Het DNA is de programmeercode van al wat leeft en bevat dus de informatie die nodig is om elk biochemisch proces in elke cel aan te sturen en al die cellen samen op te bouwen tot een volledige mens, koala, aardbei of sequoia. Het volledige DNA, uitgestrekt zo'n 2 meter lang, zit opgevouwen in de kern van elke cel van ons lichaam en wordt ook wel het 'genoom' genoemd. Het genoom bevat voor zo'n 5% code waarvan we weten dat die rechtstreeks instaat voor de productie van eiwitten. Van andere stukken wordt gedacht dat ze een regulatoire rol spelen: hoeveel, waar en wanneer worden eiwitten geproduceerd. Verder zijn er ook stukken waarvan de functie onbekend is, of is er misschien gewoon geen functie. Een groot aandeel van ons genoom bestaat uit kleine en grotere repetitieve stukken: ze komen meerdere keren voor.
Maar tussen twee mensen is er in dat genoom behoorlijk wat variatie. Deze variatie ontstaat doordat onze DNA-kopieer machinerie heel zelden in de fout gaat, maar die zeldzame fouten kunnen nadien wel worden doorgegeven aan de volgende generaties. Ook tijdens de productie van geslachtscellen kan het misgaan, wanneer stukken DNA van de ouders ongelijk worden uitgewisseld.
Het doel van genetisch onderzoek is in een speldenberg op zoek gaan naar die ene speld die scherp is en de ziekte veroorzaakt.
Opsporen van deze verschillen doen we door het volledige DNA uit te lezen, een proces dat 'sequencing' wordt genoemd. Met verschillende sequencing methodes (zie hieronder) hebben we kunnen bepalen dat het DNA van twee personen identiek blijkt voor zo'n 98.5%, met ongeveer 4.6 miljoen varianten of in totaal 47 miljoen letters verschillend. De meeste verschillen komen frequent voor in de populatie en zijn zo goed als onschadelijk en noemen we gewoon 'varianten' of 'polymorfismen'. Maar ook heel wat ziektes zijn genetisch of overerfbaar en dan spreken we van 'mutaties'. Vaak blijft het wel een moeilijk vraagstuk voor die miljoenen varianten uit te zoeken of die nu schadelijk of onschuldig zijn. Dat is het doel van genetisch onderzoek: in een speldenberg op zoek gaan naar die ene speld die scherp is en de ziekte veroorzaakt.
DNA sequencing
Sequencen van DNA gebeurt wegens technische redenen niet voor een volledig chromosoom in één stuk maar in aparte fragmenten. In 1977 werd een methode ontwikkeld waarbij die fragmenten allemaal één voor één werden uitgelezen, een heel duur en inefficiënte proces. Het was een enorm huzarenstukje, maar met deze technieken werd in 2003 het Human Genome Project afgerond, waarbij voor enkele individuen al die fragmenten in de juiste volgorde werden gepuzzeld (geassembleerd) tot een volledig humaan genoom, het referentiegenoom. Een complicatie in het assembleren zijn de repetitieve stukken van het genoom, waarbij het onduidelijk is waar deze puzzelstukjes passen.
Tegenwoordig worden de fragmenten na sequencing vergeleken met het referentiegenoom om voor elk fragment de juiste plaats te vinden, een bioinformatica taak die 'alignment' genoemd wordt. In de volgende stap kunnen dan varianten en mutaties worden geïdentificeerd door verschillen te zoeken tussen de fragmenten en de referentie. Ruwweg kunnen we de methodes voor genoom sequencing opdelen in 'korte' en 'lange' fragment sequencing. De korte fragment sequencing methodes bestaan ongeveer 15 jaar en worden op dit moment het vaakst toegepast. Bij korte fragment sequencing bekomen we erg nauwkeurige fragmenten van 50 tot 250 letters, maar deze zijn soms iets te kort om precies te aligneren met het referentiegenoom in een repetitieve regio. Lange fragment sequencing werd enkele jaren later ontwikkeld en geeft fragmenten van 10000 tot 2 miljoen letters. Hiervoor is het alignment, het zoeken van de juiste locatie in het genoom, eenvoudiger, ook bij repetitieve sequenties. Maar deze technologieën hebben als nadeel dat bij het bepalen van de nucleotiden zo'n 5-10% van de letters verkeerd is, wat de identificatie van varianten kan bemoeilijken. Deze lange fragment methodes zijn nog behoorlijk nieuw en worden daardoor veel minder toegepast in genetisch en medisch onderzoek.
DNA variatie
De meeste van die verschillen zijn slechts één letter: single nucleotide varianten (zie bijvoorbeeld Figuur 2). Een voorbeeld van een genetische ziekte door een single nucleotide variant is sikkelcelanemie, een vorm van bloedarmoede waarbij zowel het hemoglobine eiwit overgeërfd van de vader en de moeder defect is en niet goed zuurstof transporteert. Met de korte fragment sequencing, kunnen we veel van die single nucleotide varianten goed opsporen door de hoge kwaliteit van de korte fragmenten. Op dit moment is korte fragment sequencing al gedaan voor honderdduizenden patiënten of gezonde vrijwilligers. Voor genetisch onderzoek zijn deze datasets een enorme vooruitgang. Voor heel wat ziektes werden genetische oorzaken gevonden, wat dan weer belangrijk is voor diagnose en het ontwikkelen van therapieën. Helaas blijken die enorme aantallen niet voldoende om alle overerfbare ziektes op te lossen.

De zogenoemde structurele varianten zijn groter, per definitie minimaal 50 letters die veranderen van aantal of locatie, en zijn in verhouding ook veel zeldzamer. Structurele varianten komen in verschillende geuren en kleuren, zoals deleties, inversies en repeat expansies (zie Figuur 3). Ook hier weer zijn veel van deze varianten onschadelijk, maar zijn er ook die leiden tot genetische ziektes. Zo is ook een trisomie, waarbij een bepaalde chromosoom te veel voorkomt zoals in de ziekte van Down of trisomie 21, een voorbeeld van een wel erg grote structurele variant. Een ander voorbeeld van een schadelijke structurele variant is een duplicatie of zelfs triplicatie van het volledige alpha-synucleïne gen, waardoor mensen met deze variant de ziekte van Parkinson krijgen op heel jonge leeftijd.
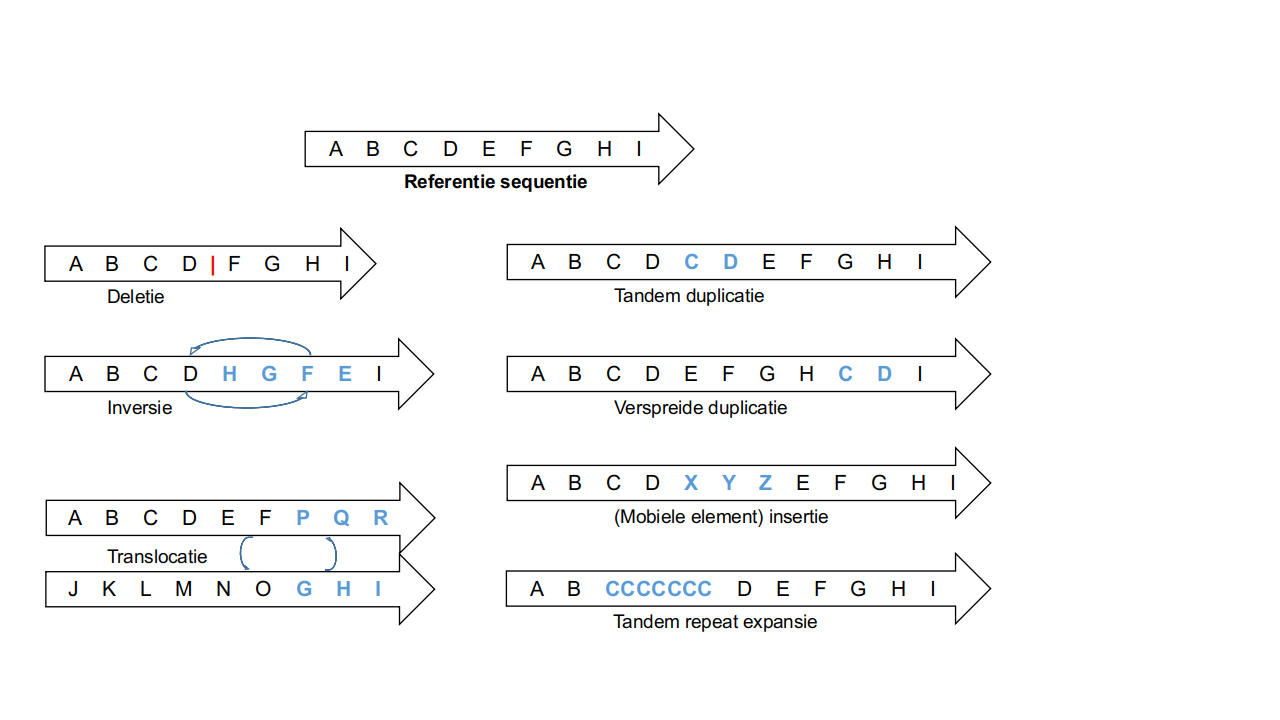
Er zijn twee redenen waardoor deze varianten te groot zijn om te vinden met korte fragment sequencing methodes. Vaak zijn deze varianten even lang of veel langer als de sequencing fragmenten, wat het alignment en variant calling behoorlijk moeilijk maakt, en structurele varianten komen juist frequent voor in repetitieve regio's in het DNA. Door die repetitieve sequenties is een correct alignment moeilijk of onmogelijk. Door deze systematische problemen zijn de meeste van de structurele varianten jaren onder de radar kunnen blijven. Lange fragment sequencing technologieën bieden daarvoor een oplossing, en met de nieuwste methodes kunnen we bepalen dat er zo'n 25 000 structurele varianten per persoon kunnen gevonden worden. Dat is samen goed voor 37 miljoen letters die verschillend zijn, en hierdoor hebben deze zeldzame varianten meteen ook de grootste invloed op de variatie tussen twee mensen, en zeer waarschijnlijk ook een rol in het ontstaan van ziektes. In mijn onderzoeksproject gaan we op zoek naar structurele varianten met een rol in neurodegeneratieve hersenziekten, zoals de ziekte van Alzheimer en Frontotemporale Dementie. Het toepassen van lange fragment sequencing op meer individuen zal de komende jaren hopelijk onze kennis van vele genetische aandoeningen gevoelig vergroten.