Twintig jaar, dat is de tijd tussen de ontdekking van een nieuw materiaal en wanneer het in onze rekken ligt. We kunnen dit proces versnellen door machine learning modellen te trainen om de meest veelbelovende moleculen op te sporen. Net zoals een baasje zijn speurhond traint.
Wetenschappers spenderen ongeveer 20 jaar aan onderzoek om van een fundamentele ontdekking naar een bruikbaar materiaal te gaan. Maar waarom wachten we hier zo lang op? Zijn wetenschappers niet te lui, of werken ze niet te traag? Die stelling kunnen we betwisten. Zo hebben chemici tot op heden meer dan 196 miljoen moleculen ontdekt en gemaakt, waarvan enkele zo’n grote impact hebben op onze maatschappij dat we haast vergeten dat ze er zijn.
196 miljoen, een impressionante hoeveelheid, maar als we dit getal van een afstand bekijken en realiseren dat er 10 tot de macht 60 unieke kleine moleculen zijn − dat is trouwens een pak meer dan het aantal zandkorrels op onze aardbol − dan pas komt het besef dat we als wetenschappers nog een lange weg af te leggen hebben. In recent onderzoek toonden we aan dat we met behulp van computersimulaties en machine learning, meer gericht kunnen zoeken naar nuttige moleculen en zo sneller die ene naald in de hooiberg kunnen opsporen. Onze case - de supramoleculaire gel.
Gellen voor cellen
De supramoleculaire gel is een materiaal dat zijn intrede maakte in laboratoria in het begin van de 21ste eeuw. De interesse in dit materiaal kan verklaard worden door de gelijkenissen die het heeft met de extracellulaire matrix, een weefsel dat aanwezig is in ons lichaam en structuur biedt aan cellen. De extracellulaire matrix is dus noodzakelijk om de celgroei te bevorderen. Stel dat we dan het tekort aan donororganen zouden willen aanpakken door deze zelf te kweken in het laboratorium, dan is het ook van cruciaal belang dat we een alternatief hebben voor die extracellulaire matrix. Met andere woorden, een perfect geoptimaliseerde supramoleculaire gel.
Wetenschappers kunnen niet voorspellen of de gel die ze maken de juiste eigenschappen zal vertonen
Om een supramoleculaire gel te maken hebben we twee bestandsdelen nodig: een vloeistof en kleine organische moleculen. Het unieke aspect van de supramoleculaire gel zijn de reversibele interacties die de kleine moleculen met elkaar aangaan in de vloeistof. Hierdoor klitten ze samen en vormen ze één groot super-netwerk van miljoenen kleine moleculen. Zo’n interacties noemen we binnen de chemie supramoleculaire bindingen, vandaar de naam supramoleculaire gel. Helaas kunnen wetenschappers momenteel niet voorspellen of de eigenschappen van de supramoleculaire gel die ze maken, geschikt zal zijn voor de toepassing die ze voor ogen hebben. Meer nog, de kleinste aanpassing aan de organische molecule resulteert vaak in het volledige verlies van de interacties die de gel net zo interessant maakt. Dit heeft als gevolg dat de huidige strategie voor de optimalisatie en ontwikkeling van de supramoleculaire gel een tijdrovend proces is, en we als chemici vaak met onze handen in het haar zitten.

Kleine moleculen vormen een netwerk in een supramoleculaire gel.
Een computer begeleidt ons naar de juiste molecule
In onderzoek uitgevoerd aan de KU Leuven en de Vrije Universiteit Brussel willen wij het optimalisatie proces versnellen door het geweer over een andere schouder te gooien. In plaats van een reeks experimenten op te starten proberen we eerst het gedrag van de gel beter te begrijpen door middel van virtuele simulaties. In deze simulaties plaatsen we de kleine moleculen ver weg van elkaar en volgen we op of ze de neiging hebben om aan elkaar te gaan klitten. Niet alleen dit, maar we bestuderen ook hoe ze exact aan elkaar klitten en of de gevormde structuur zou kunnen leiden tot een geschikt netwerk. Dit deden we aan de hand van descriptoren, getallen die een waarde geven aan hoe graag de moleculen samen klitten. Door deze descriptoren op hun beurt in te voeren in een machine learning model, zoals bijvoorbeeld een neuraal netwerk, kunnen we dan op voorhand voorspellen of een molecule al dan niet in staat is een gel te vormen. In feite gebruiken we dus de simulaties in combinatie met machine learning modellen als een soort gel-detector en kunnen we hierdoor onze tijd in het laboratorium een pak efficiënter spenderen.
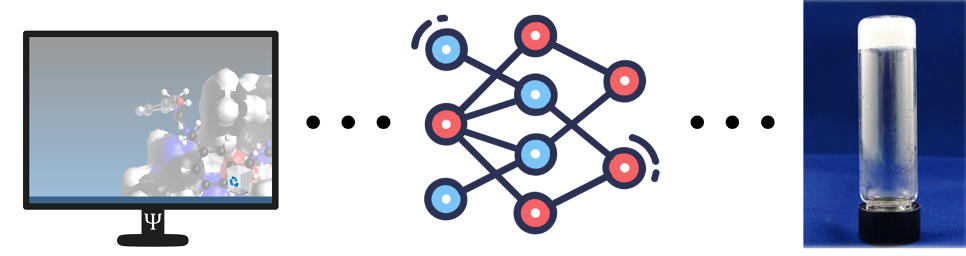
Computer simulaties geven aanleiding tot een machine learning model waardoor we sneller een supramoleculaire gel kunnen ontdekken.
In 2018 kondigde MIT aan 1 miljard dollar te investeren in opleidingen gericht op artificiële intelligentie
Computers worden meer en meer gebruikt om sneller nuttige moleculen te ontdekken in verschillende disciplines. Zo wordt de afgelopen jaren machine learning ook toegepast om nieuwe medicijnen te ontdekken of biologisch afbreekbare plastics. Geen wonder dat de Massachutes Institute of Technology (MIT) in de Verenigde Staten in 2018 aankondigde maar liefst 1 miljard dollar te investeren in opleidingen gericht op artificiële intelligentie. Wil dit dan zeggen dat de job chemist gedoemd is uit te sterven? Niet bepaald, want hoewel machine learning ons in staat stelt om sneller nuttige moleculen te ontdekken, werkt deze strategie alleen als de modellen goed getraind zijn. Vergelijk het met een speurhond, die enkel zijn job zal kunnen uitvoeren als het baasje hem goed heeft opgevoed. Het is dus aan ons wetenschappers om in de komende jaren uit te dokteren hoe we deze machine learning modellen het beste kunnen “opvoeden” om zo de juiste moleculen op te sporen. En hiervoor hebben we onze chemische intuïtie nog steeds hard nodig.
Ruben Van Lommel dingt mee naar de Vlaamse PhD Cup 2022. Ontdek meer over dit onderzoek op www.phdcup.be.